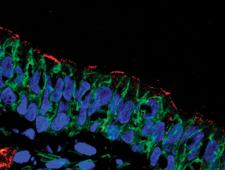
David Stoltz, MD, PhD, and colleagues study the pathogenesis of cystic fibrosis, a multisystem genetic disease that begins with the dysfunction of an anion channel, called the cystic fibrosis transmembrane conductance regulator (CFTR).
As the disease progresses, thick mucus accumulates in a number of organs—including the gallbladder. This mucus obstruction interferes with the function of the gallbladder, to store and secrete bile, and it may also be involved in additional hepatobiliary complications. Studying the gallbladder may give insight into how dysfunction of CFTR leads to abnormal mucus accumulation.
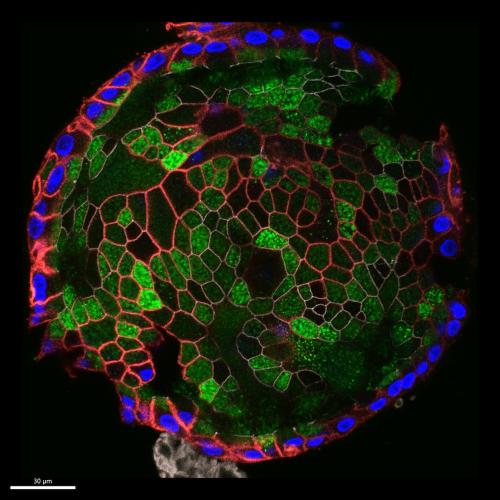
The image, submitted by graduate student Keyan Zarei, is a human gallbladder epithelial organoid (a mini-representation of an organ, grown in a dish). The organoids form tiny spheres (50-200 micrometers in diameter), where a layer of cells surrounds a fluid-filled lumen.
The blue portions of the image highlight the nuclei of the organoid cells. The red portions label an ion pump that is localized at the bottom of the cell. The green areas mark MUC5AC, a protein that makes up a part of the problematic mucus.